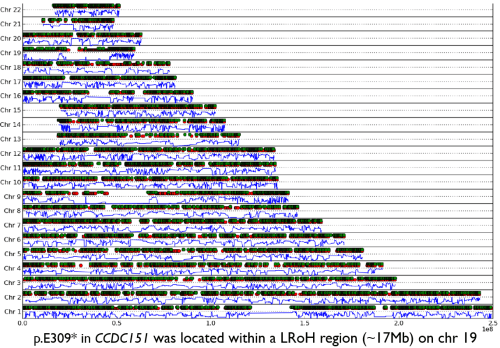
An example output from AutoZplotter using whole-exome sequencing data – used to identify the Primary ciliary dyskinesia causal gene, CCDC151, in Alsaadi and Erzurumluoglu et al, 2014 (read this paper’s story here). The green and red dots correspond to heterozygous and homozygous calls (for the alternative allele), respectively. The continuous blue lines correspond to the probability that the observed sequence of genotypes is not autozygous (e.g. close to zero means likely to be an autozygous region). LRoH: Long runs of homozygosity. NB: This image has been edited to ensure confidentiality/anonymity of the participant. Some LRoHs have been shortened or extended for this reason. If you’re thinking of using an AutoZplotter image in a paper, do not share genome-wide figures but maybe consider using chromosome-wide ones
When analysing whole-exome or whole-genome sequencing (or dense SNP chip) data obtained from consanguineous individuals with a rare Mendelian disease, the disease causal mutation usually lies within an autozygous region (characterised by long runs of homozygosity, LRoH, which are generally >5Mb). Thus checking whether any candidate genes overlap with an LRoH can substantially narrow region(s) of interest. There are several tools which can identify LRoHs such as Plink, AutoSNPa and AgilentVariantMapper. However, they all require their own formats and considerable computational knowledge; and also struggle to identify regions that are shorter than 5Mb. Thus, we wrote AutoZplotter, a user-friendly python script which plots the heterozygosity/homozygosity status of variants in a VCF file to allow for quick visualisation and manual identification of regions that have longer stretches of homozygosity than would be expected by chance.

AutoZplotter accepts the VCF format – which is the standard format for storing genetic variation data from NGS platforms. Image Source URL: bioinf.comav.upv.es
The input format of AutoZplotter is VCF, thus it will be suitable for any type of genetic data (e.g. SNP array, WES, WGS) and from any species.
An older version of AutoZplotter was used in the analysis stage of Alsaadi et al (2012) and Alsaadi and Erzurumluoglu et al (2014).
To download latest version of AutoZplotter, click here (directs to ResearchGate). If you found AutoZplotter helpful in anyway, please cite Erzurumluoglu AM et al, 2015.
References:
Erzurumluoglu AM et al, 2015. Identifying Highly Penetrant Disease Causal Mutations Using Next Generation Sequencing: Guide to Whole Process. BioMed Research International. Volume 2015 (2015), Article ID 923491
Alsaadi MM and Erzurumluoglu AM et al, 2014. Nonsense Mutation in Coiled-Coil Domain Containing 151 Gene (CCDC151) Causes Primary Ciliary Dyskinesia. Human Mutation. Volume 35, Issue 12. Pages 1446–1448
Erzurumluoglu AM et al, 2016. Importance of Genetic Studies in Consanguineous Populations for the Characterization of Novel Human Gene Functions. Volume 80, Issue 3. Pages 187–196
Erzurumluoglu AM, 2015. Population and family based studies of Consanguinity: Genetic and Computational approaches. PhD Thesis. University of Bristol




