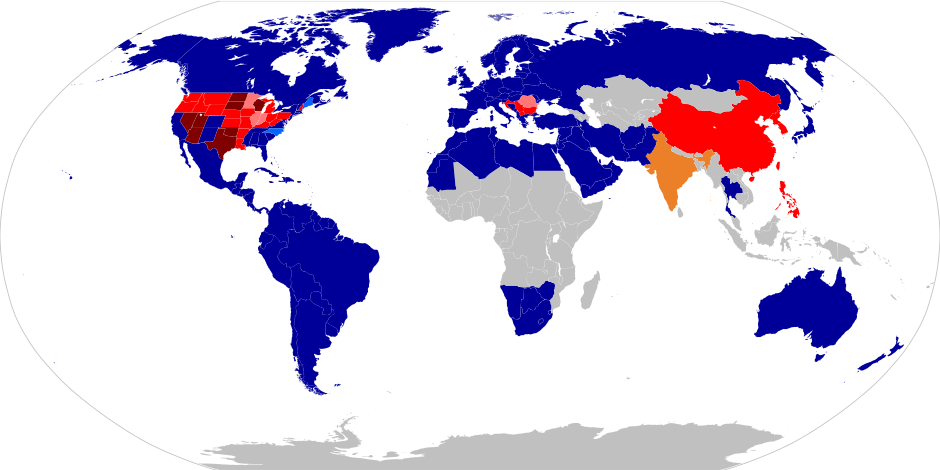
Laws regarding first-cousin marriage around the world. Navy blue: First-cousin marriage legal. Light Blue: Allowed with restrictions or exceptions. Yellow: Legality dependent on religion or culture. Red: Statute bans first-cousin marriage. Pink: Banned with exceptions. Dark Red: Criminal offense. Grey: No available data. The image has been released into the public domain by the author (URL: http://en.wikipedia.org/wiki/Cousin_marriage).
The answer is (studying) consanguinity (i.e. unions between relatives such as first-cousin marriages); and one cannot understand the complexity of the issue (and make ‘informed’ decisions) without reading the literature of these five apparently unconnected fields. It is fair to say that there is a degree of hostility towards consanguineous marriages in Western societies. However this perception is usually attained without in-depth knowledge on the genetic effects of consanguinity. In short, consanguinity per se (i.e. on its own) does not cause a disorder, but rather it increases the probability of an autosomal recessive disorder (which require two copies of the same) causal mutation to be in a homozygous state (i.e. possess two copies of the same mutation). When this happens both copies of the genes we inherited from our parents do not function properly.
Unions between individuals who are second-cousins or closer are considered ‘consanguineous’ in clinical genetics. Consanguineous families with diseases have been studied thoroughly by clinical geneticists for the last two-three decades – and this has allowed for identification of many disease causal genes. However, studying consanguineous populations as a whole rather than ‘cherry picking’ families with disease can offer much more for better understanding our genome and therefore finding new targets for preventive and curative medicine. Many genes in our genome still have unknown functions and we have merely scratched the surface in terms of their interactions. I hypothesise that assigning a function to the thousands of remaining genes will only be feasible if consanguineous populations are studied as a whole (i.e. also including families without disease to the studies) and I therefore carry out theoretical studies to estimate the sample size needed and how many genes will be completely ‘knocked-out’ if these studies were to be carried out. This approach proposes a ‘paradigm shift’ in clinical genetics.
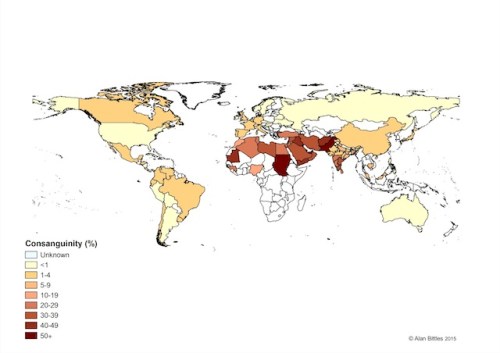
Global prevalence of consanguineous unions. Consanguinity has deep roots in many cultures and it is impossible to interfere/intervene from the outside without first understanding why people engage in cousin marriages. Image source URL: http://www.consang.net/
Consanguineous unions occur very rarely in Western countries for a variety of sociological (e.g. cultural, negative media coverage) and statistical reasons (e.g. smaller families means fewer cousins at similar age), but the complete opposite is true in certain regions of the world where union of kin is seen as the default choice. Conservative estimates predict that approximately one-sixth of the world’s population (a figure of 1.1 billion is proposed by the Geneva International Consanguinity Workshop Report) live in highly consanguineous regions; and also another one-sixth falls into the ‘unknown’ category – reflecting the need for further research. Historically, consanguineous unions were also common amongst the elite in the UK (up to mid-19th century, including Charles Darwin), the Pharaohs and the Royal families of Europe (e.g. Habsburgs).

Views of main religions towards consanguineous marriages. NB: Where first-cousin marriages are allowed, lower levels of consanguinity are also allowed. Image Source: Copy-pasted from my own PhD thesis
The increase in the probability of a mutation being homozygous will depend on the level of relatedness between the parents. For example, approximately 6.25% of mutations are expected to be homozygous in the offspring of first cousins. This figure would be (near) 0% in the offspring of outbred individuals. Genetically, this is the main difference between union of kin and union of unrelated individuals. We all have many disease-causal mutations in our genomes (but in heterozygous state, i.e. one normal copy and one mutated copy) and different kinds of mutations are out there in all populations. However because these mutations will be very rare or are unique to you or your family, they do not get to meet their counterpart when you have offspring with an unrelated individual. Therefore the mutation’s homozygous effects are never observed. This is why rare autosomal recessive disorders are almost always seen in consanguineous offspring.
This difference in homozygosity levels is also one of the main reasons behind the necessity of studying consanguineous individuals and populations. These studies can turn unfortunate events (e.g. disorder in families) to a great use for medical sciences. Not only will identifying a disease-causal mutation help with diagnostics, they can enable scientists to understand what certain genes do and help us understand why the gene causes that disease. Rare instances can be highly informative about preventable outcomes relevant to the whole population. For example, had more notice been taken in the 1980s of the proof which familial hypercholesterolemia provided for the causal role of cholesterol in coronary heart disease (CHD), high cholesterol intake would have been better addressed for the nation a decade sooner. To provide numbers, CHD is still the UK’s biggest killer causing over 80 thousand deaths a year, thus paying more attention to information that was coming from studies of consanguineous unions could have saved thousands of lives just in this single case.
Given the advancements in genetic diagnostics (e.g. huge decreases in costs of DNA sequencing), screening for all known mutations will become feasible in the near future for everybody – and identifying disease-causal mutations will become even more useful for all of us. Our genomes are constantly being mutated and my approach will enable a much broader understanding of our genome by observing these mutations’ homozygous effects. Finally, rather than discourage (See link for an example) consanguineous marriages totally (not feasible in the foreseeable future due to many socio-economic and cultural reasons), for those willing to marry a cousin, screening for previously identified mutations will help these couples take more informed decisions.

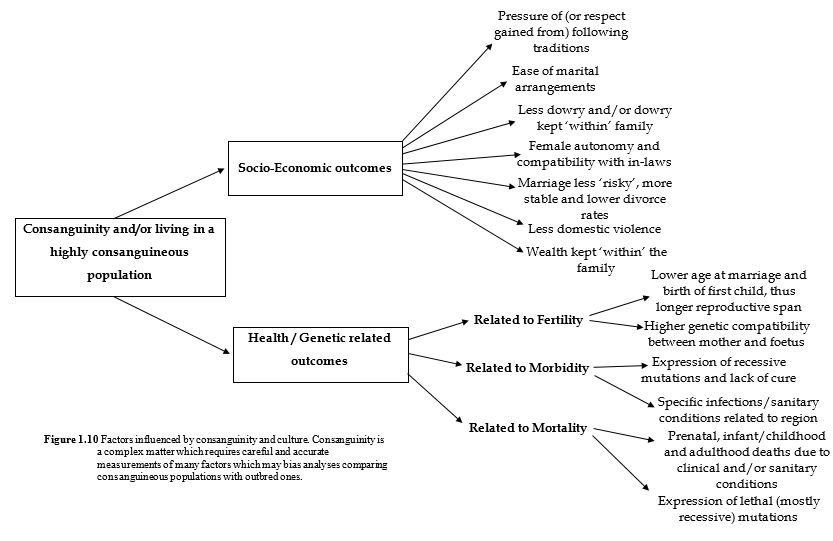
Factors influenced by consanguinity and culture. Image Source: Copy-pasted from my own PhD thesis (hence the Figure 1.10)
Key reference:
A. Mesut Erzurumluoglu, 2016. Population and family based studies of consanguinity: Genetic and Computational approaches. PhD thesis. University of Bristol.
Erzurumluoglu AM et al, 2016. Importance of Genetic Studies in Consanguineous Populations for the Characterization of Novel Human Gene Functions. Annals of Human Genetics, 80: 187–196.
PS: Whilst the media is mostly responsible for portraying consanguinity the way they understand (and with more contrast added on of course), they could be forgiven as the genetic effects of consanguinity is not fully understood amongst geneticists either, especially in the field of complex trait genetics – thus the extra incentive for studying them.
Read Full Post »